氨基酸全序列比对
生物学家劳伦斯·赫斯特和斯蒂芬·弗里兰在20世纪90年代末把天然基因密码和计算机随机产生的几百万组密码拿去比对,结果轰动一时。他们想知道,如果发生点突变这种把一个字母换掉的变异,哪一套密码系统能保留最多正确的氨基酸,或将它代换成另一个性质相似的氨基酸。
结果他们发现,天然的基因密码最经得起突变的考验。点突变常常不会影响氨基酸序列,而如果突变真的改变了氨基酸,也会由另一个物理特性相似的氨基酸来取代。据此,赫斯特与弗里兰宣称,天然的遗传密码比成千上万套随机产生的密码要优良得多。它不但不是大自然密码学家愚蠢而盲目的作品,而是万里挑一的密码系统。
天然的三联基因密码的第一个字母都有特定的对应方式。举例来说,所有以丙酮酸为前体合成的氨基酸,它们密码的第一个字母都是T。所有由α-酮戊二酸所合成的氨基酸,其三联密码第一个字母都是C;所有由草酰乙酸合成的氨基酸,第一个字母都是A;最后,几种简单前体通过单一步骤所合成的氨基酸,第一个字母都是G。
三联密码的第二个字母和氨基酸是否容易溶于水有关,或者说和氨基酸的疏水性有关。亲水性氨基酸会溶于水,疏水性氨基酸不会溶于水,但会溶在脂肪或油里,比如溶在含有脂质的细胞膜里。所有的氨基酸,可以从“非常疏水”到“非常亲水”排列成一张图谱,而正是这张图谱决定了氨基酸与第二个密码字母之间的关系。疏水性最强的六个氨基酸里有五个,第二个字母都是T,所有亲水性最强的氨基酸第二个字母都是A。介于中间的有些是G有些是C。
三联密码的第三个字母不含任何信息,不管接上哪一个字母都没关系,这组密码子都会翻译出一样的氨基酸。以甘氨酸为例,它的密码子是GGG,但是最后一个G可以代换成T、A或C。
第三个字母的随机性暗示了一些有趣的事情。二联密码可以编码16种氨基酸。如果我们从20个氨基酸里拿掉5个结构最复杂的(剩下15个氨基酸,再加上一个终止密码子)这样前两个字母与这15个氨基酸特性之间的关联就更明显了。因此,最原始的密码可能只是二联密码,后来才靠“密码子捕捉”的方式成为三联密码,也就是各氨基酸彼此竞争第三个字母。
第一个字母和氨基酸前体之间的关系直截了当,第二个字母和氨基酸的疏水性相关,第三个字母可以随机选择。这套密码系统除了可以忍受突变,还可以降低灾难发生时造成的损失,同时可以加快进化的脚步。因为如果突变不是灾难性的,那应该会带来更多的好处。
氨基酸序列决定蛋白质三维结构
作为一种具有“推动者”(Enabler)地位的先进技术,人工智能在各大科学领域都有着极大的应用潜力,尤其是生物医疗领域。近日,麻省理工学院的研究人员通过机器学习模型分析氨基酸序列,在无需其他任何蛋白质结构信息作为输入数据下,成功的掌握了蛋白质的三维结构,并进一步的预测其生物学功能。
这项研究将在今年 5 月的国际学习表征会议(ICLR)上发表,相关论文目前正在收集评论,其成果有望改进现有的蛋白质结构预测手段,帮助科学家设计和测试新蛋白质,用于药物研发和生物研究等目的。
蛋白质是由氨基酸组成的线性链,且每一种都有自己独特的氨基酸序列,通过肽键连接。根据氨基酸的序列和物理相互作用,蛋白质分子可以折叠成非常复杂的三维结构,而这种结构决定了其生物学功能,比如对药物产生什么样的反应。
图 | 蛋白质分子的三维模型 (来源:MIT News)
然而,经过数十年的研究和多种成像技术的发明,人们仍然只掌握了很少一部分蛋白质的结构——在已知的上百万种蛋白质中,我们只了解其中几万个的结构。
鉴于氨基酸序列的信息相对比较容易获取,麻省理工学院的研究人员想到了擅长预测和寻找规律的机器学习技术,希望可以组合不同的氨基酸序列,从而找到新的蛋白质结构。这是一个富有挑战性的任务,因为不同的氨基酸序列可以形成非常相似的结构,而且没有很多结构可以用来训练模型。
研究的第一作者 Tristan Bepler 认为,类似的研究可以将蛋白质结构的预测边缘化,因为只需要氨基酸序列,就可以推测出蛋白质的功能。
预测蛋白质结构
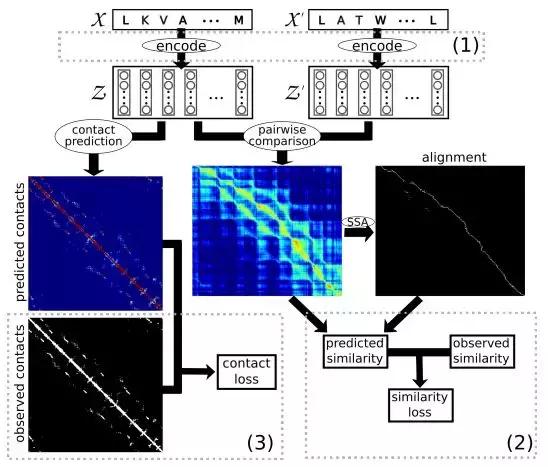
研究团队没有直接基于蛋白质结构建立预测模型,而是首先尝试编码蛋白质的结构信息,将其变为一种易于计算的表达方式,再训练模型学习特定氨基酸的功能,找出不同蛋白质结构之间的相似度,然后用这一数据来监督模型。
依据蛋白质结构分类数据库(SCOP)的数据,研究人员对大约 22,000 种蛋白质进行了模型训练,通过其结构和氨基酸序列的相似性分成不同类别。
他们随后将蛋白质结构和氨基酸序列编码,转换成套嵌(Embedding)的数字表达形式,以随机配对的形式放入预测模型中,每组套嵌包括两个氨基酸序列的相似性信息,经过对比和计算,可以得出蛋白质三维结构的相似度,最后根据其中每个氨基酸的位置和接触来预测其功能。
图 | 模型的工作过程 (来源:Tristan Bepler)
我们可以用类比搞清楚它的运作机制:在自然语言处理(NLP)的语义分析中,也存在类似的套嵌概念,比如两套单词的套嵌(值)越相近,它们在句子中一起出现的可能性就越大。
类比到蛋白质结构预测模型中,蛋白质就像“句子”,氨基酸就像“单词”,那么氨基酸序列也就成为了“单词的组合形式”,而套嵌则是“单词组合的相似度”,可以对比得出“句子(蛋白质)的相似度”。如果两组氨基酸序列越相近,它们的套嵌(值)就越相近,就说明两种蛋白质结构越相似。
在训练过程中,机器学习模型负责计算两组套嵌的相似性得分,然后预测出蛋白质三维结构的相似性,再与实际 SCOP 相似性得分进行比较。如果两者非常接近,就说明模型的方法没问题,反之则需要调整。
此外,该模型还会预测每组套嵌的“接触图”(Contact Map),即三维结构中每个氨基酸与其他氨基酸的距离。这有助于模型掌握氨基酸在蛋白质结构中的确切位置,从而进一步预测每种氨基酸的功能。同样的,它会跟 SCOP 数据中已知的接触图进行比较,验证判断是否准确。
作为测试,研究人员利用该模型预测哪些蛋白质可以穿过细胞膜,在仅给出一个氨基酸序列的情况下,所得结果的准确率已经超过了现有的最先进的模型。
接下来,研究团队的目标是将该模型应用于更多预测任务,拓展到更广泛的蛋白质工程领域,例如检测破坏蛋白质结构的有害突变,以及确定哪些氨基酸序列片段可以与小分子结合,这对于药物研发至关重要。该模型还可以用于蛋白质设计领域,通过对套嵌等数据的分析,可以找出更多使蛋白质发光的波长。
“机器学习模型可以有效利用已知的氨基酸序列,分析蛋白质折叠的奥秘,进而推测未知的蛋白质结构和功能。我们的终极目标是,更有效地赋能数据驱动的蛋白质设计工程。” Bepler 强调。
分析氨基酸序列,AI 撬开未知蛋白质功能大门,氨基酸序列决定蛋白质三维结构
四种DNA字母要编码20种氨基酸。绝不可能是一对一编码,也不可能是二对一编码,因为两个字母最多只能组成16种组合(4×4)。因此,最低要求是三个字母,也就是DNA序列里面最少要有三个字母对应到一个氨基酸,被称为三联密码,后来被克里克和西德尼·布伦纳证实。
但是这样看起来似乎很浪费,因为用四种字母组成三联密码,总共可以有64种组合(4×4×4),这样应该可以编码64个不同的氨基酸,那为什么只有20种氨基酸呢?一定有一个神奇的答案来解释为什么4种字母,3个一组,拼成64个单词,然后编码20种氨基酸。
1952年,沃森就曾写信告诉克里克:“DNA合成信使RNA(mRNA), mRNA合成蛋白质。”克里克开始研究这一小段mRNA的字母序列,如何翻译成蛋白质里面的氨基酸序列。他认为mRNA可能需要一系列“适配器”来帮助完成翻译,每一个适配器都负责携带一个氨基酸。当然每一个适配器一定也是RNA,而且都带有一段“反密码子”序列,这样才能和mRNA序列上的密码子配对。
适配器分子也由RNA分子组成。它们现在叫作“转运RNA”或tRNA。现在整个工程变得有点像乐高积木,一块块积木接上来又掉下去,一切顺利的话,它们就会这样一个接一个地搭成精彩万分的聚合物。
随着实验技术进步而且越来越精密,在20世纪60年代中期许多实验室陆续解开了序列密码。然而经过一连串不懈的译码工作后,大自然却好像随兴地给了个潦草结尾,让人既困惑又扫兴。遗传密码子的安排一点也不具创意,只不过“简并”了(意思就是说,冗余)。有三种氨基酸可对应六组密码子,其他的则各对应一到两组密码子。每组密码子都有意义,还有三组的意思是“在此停止”,剩下的每一组都对应一个氨基酸。这看起来既没规则也不美,根本就是“美是科学真理的指南”这句话的最佳反证。甚至,我们也找不出任何结构上的原因来解释密码排列,不同的氨基酸与其对应的密码之间似乎并没有任何物理或化学的关联。
克里克称这套让人失望的密码系统为“冻结的偶然”,而大部分人也只能点头同意。他说这个结果是冻结的,因为任何解冻(试图去改变密码对应的氨基酸)都会造成严重的后果。一个点突变也许只会改变几个氨基酸,而改变密码系统本身却会从上到下造成天大灾难。就好似前者只是一本书里无心的笔误,并不会改变整本书的意义,然而后者却将全部的字母转换成毫无意义的乱码。克里克说,密码一旦被刻印在石板上,任何想改动它的企图都会被处以死刑。这个观点至今仍有许多生物学家认同。



 添加微信免费咨询
添加微信免费咨询
