蛋白质氨基酸序列的测定
四种DNA字母要编码20种氨基酸。绝不可能是一对一编码,也不可能是二对一编码,因为两个字母最多只能组成16种组合(4×4)。因此,最低要求是三个字母,也就是DNA序列里面最少要有三个字母对应到一个氨基酸,被称为三联密码,后来被克里克和西德尼·布伦纳证实。
但是这样看起来似乎很浪费,因为用四种字母组成三联密码,总共可以有64种组合(4×4×4),这样应该可以编码64个不同的氨基酸,那为什么只有20种氨基酸呢?一定有一个神奇的答案来解释为什么4种字母,3个一组,拼成64个单词,然后编码20种氨基酸。
1952年,沃森就曾写信告诉克里克:“DNA合成信使RNA(mRNA), mRNA合成蛋白质。”克里克开始研究这一小段mRNA的字母序列,如何翻译成蛋白质里面的氨基酸序列。他认为mRNA可能需要一系列“适配器”来帮助完成翻译,每一个适配器都负责携带一个氨基酸。当然每一个适配器一定也是RNA,而且都带有一段“反密码子”序列,这样才能和mRNA序列上的密码子配对。
适配器分子也由RNA分子组成。它们现在叫作“转运RNA”或tRNA。现在整个工程变得有点像乐高积木,一块块积木接上来又掉下去,一切顺利的话,它们就会这样一个接一个地搭成精彩万分的聚合物。
随着实验技术进步而且越来越精密,在20世纪60年代中期许多实验室陆续解开了序列密码。然而经过一连串不懈的译码工作后,大自然却好像随兴地给了个潦草结尾,让人既困惑又扫兴。遗传密码子的安排一点也不具创意,只不过“简并”了(意思就是说,冗余)。有三种氨基酸可对应六组密码子,其他的则各对应一到两组密码子。每组密码子都有意义,还有三组的意思是“在此停止”,剩下的每一组都对应一个氨基酸。这看起来既没规则也不美,根本就是“美是科学真理的指南”这句话的最佳反证。甚至,我们也找不出任何结构上的原因来解释密码排列,不同的氨基酸与其对应的密码之间似乎并没有任何物理或化学的关联。
克里克称这套让人失望的密码系统为“冻结的偶然”,而大部分人也只能点头同意。他说这个结果是冻结的,因为任何解冻(试图去改变密码对应的氨基酸)都会造成严重的后果。一个点突变也许只会改变几个氨基酸,而改变密码系统本身却会从上到下造成天大灾难。就好似前者只是一本书里无心的笔误,并不会改变整本书的意义,然而后者却将全部的字母转换成毫无意义的乱码。克里克说,密码一旦被刻印在石板上,任何想改动它的企图都会被处以死刑。这个观点至今仍有许多生物学家认同。
谷歌AI一次注释了10%的已知蛋白质序列,超过人类十年研究成果
机器之心报道
编辑:泽南、张倩
和 AlphaFold 不同,这次谷歌探索的是用深度学习给蛋白质打上功能标签。
蛋白质是组成人体一切细胞、组织的重要成分。机体所有重要的组成部分都需要有蛋白质的参与。
目前已知存在的蛋白质种类有数十亿,但其中大约有三分之一的功能是不可知的。我们迫切地需要探索这片未知区域,因为它们关系到抗菌素耐药性,甚至气候变化等重要议题。例如,青霉素是蛋白质之间自然反应的产物,植物蛋白可用于减少大气中的二氧化碳。
近日,谷歌与欧洲生物信息学研究所合作开发了一种技术 ProtCNN,其能够使用神经网络可靠地预测蛋白质功能,帮助我们缩小蛋白质宇宙中最后不可见的区域。
谷歌表示,这种新方法让我们可以较为准确地预测蛋白质功能、突变的功能效应,并进行蛋白质设计,进而应用于药物发现、酶设计,甚至是了解生命的起源。
论文:Using deep learning to annotate the protein universe
论文链接:https://www.nature.com/articles/s41587-021-01179-w
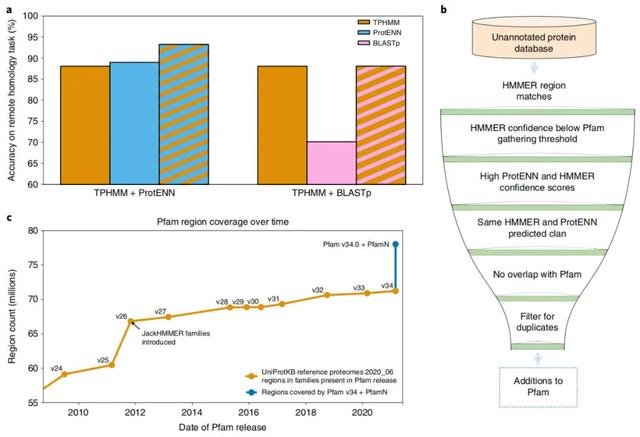
谷歌提出的方法可靠地预测了更多蛋白质的作用,而且它们快速、便宜且易于尝试,其研究已让主流数据库 Pfam 中注释的蛋白质序列增加了近 10%,一举超过了过去十年的增速,并预测了 360 种人类蛋白质功能。
Pfam 数据库是一系列蛋白质家族的集合,其中每一个蛋白家族都以多序列比对和隐马尔科夫模型的形式来表示。
这些结果表明,深度学习模型将成为未来蛋白质注释工具的核心组成部分。
对于大多数人来说,我们更熟悉的是 DeepMind 此前预测蛋白质结构算法 AlphaFold 的工作。AlphaFold 向我们展示了这些神秘生物机器的形状,新研究的重点则是这些机器的作用以及它们的用途。
生物医疗是一个极其活跃的科学领域,每天都有超过十万个蛋白质序列被添加到全球序列数据库中。但是,除非附有功能注释,否则这些条目对从业者的用途非常有限。虽然人们会努力从文献中提取注释,每年评估超过六万篇论文,但这项任务的耗时性质意味着只有 0.03% 的公开可用蛋白质序列是手动注释的。
直接从氨基酸序列推断蛋白质功能是科学社区长久以来一直在研究的方向。从 1980 年代开始,人们就提出了 BLAST 等方法,其依赖于成对的序列比较,假设查询蛋白与已经注释的高度相似的序列具有相同的功能。后来,人们引入了基于 signature 的方法,PROSITE 数据库对在具有特定功能的蛋白质中发现的短氨基酸「基序」进行分类。基于 signature 方法的一个关键改进是开发了 profile 隐马尔可夫模型(pHMM)。这些模型将相关蛋白质序列的对齐折叠成一个模型,该模型为新序列提供似然分数,描述它们与对齐的集合的匹配程度。
在这里至关重要的是,profile HMM 允许更长的 signature 和更模糊的匹配,目前用于更新流行的数据库,如 Interpro 和 Pfam。后期的改进使这些技术更加灵敏,计算效率更高,而它们作为网络工具的高可用性让从业者可以轻松将它们整合到工作流程中去。
这些计算建模方法在学界产生了很大影响。然而,至今仍有三分之一的细菌蛋白质没有被注释出功能。究其原因,当前方法对每个比较序列或模型进行完全独立的比较,因此可能无法充分利用不同功能类共享的特征。
扩展注释的蛋白质序列集需要远程同源检测,即对与训练数据相似度低的序列进行准确分类。新研究得到的基准测试集包含 21,293 个序列。ProtENN 对所有类别分类的准确度显着提高,包括那些具有远距离测试序列的类,这是扩大蛋白质领域覆盖范围的关键要求。为解决从几个例子中推断的挑战,作者使用深度模型学习的序列表示来提高性能。
Pfam-seed 模型的性能。
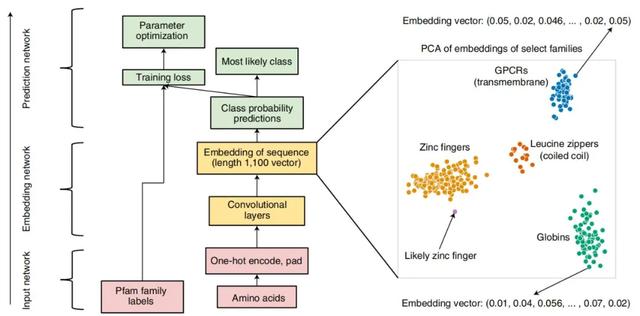
ProtCNN 的架构。中心图展示了输入(红色)、嵌入(黄色)和预测(绿色)网络以及残差网络 ResNet 架构(左),而右图展示了 ProtCNN 和 ProtREP 通过简单的最近邻方法利用。在这一表示中,每个序列对应一个点,来自同一家族的序列通常比来自其他家族的序列更接近。
ProtCNN 学习每序列长度为 1100 的实值向量表示,无论其未对齐长度如何。为获得高精度,来自每个族的表示必须紧密地聚集在一起,以便不同的族很好地相互分离。为了测试这种学习表示是否可用于准确分类最小家族的序列,作者构建了一种称为 ProtREP 的新方法。对于 ProtREP,研究者计算每个家族在其训练序列中的平均学习表示,产生一个标记家族表示。然后通过在学习表示空间中找到其最近的标记来对每个保留的测试序列进行分类。对于相同的计算成本,ProtREP 在聚类分割上超过了 ProtCNN 的准确性。
ProtENN 和 TPHMM 的组合提高了远程同源任务的性能。TPHMM 和 ProtENN 模型的简单组合将错误率降低了 38.6%,将 ProtENN 数据的准确度从 89.0% 提高到 93.3%。
为探究深度模型对蛋白质序列数据的了解,作者在来自 Pfam-full 的 80% 的未对齐序列上训练 ProtCNN,并计算了学习氨基酸表示的相似性矩阵。
结果表明,ProtCNN 学习了一种有意义的蛋白质序列表示方式,其可泛化到序列空间未知的部分,可用于预测和理解蛋白质序列的特性。另一个挑战是检测蛋白质结构域及其在蛋白质序列中的位置。此任务类似于图像分割,这正是深度学习模型擅长的任务。虽然 ProtCNN 是使用域进行训练的,但研究展示了 ProtCNN 使用简单的滑动窗口方法将完整序列分割成域的能力。
尽管不使用序列比对,但 ProtCNN 仍显示出了卓越的准确性。
谷歌AI一次注释了10%的已知蛋白质序列,超过人类十年研究成果
#科学燃计划##科学前沿#【AlphaFold精准预测蛋白结构】
蛋白质三维结构预测是生物学最严峻的挑战之一。继围棋、国际象棋等竞技项目之后,近日谷歌旗下DeepMind开发的人工智能程序AlphaFold在两年一次的蛋白质结构预测挑战赛CASP中再次大幅胜出。该程序在根据蛋白质氨基酸序列确定蛋白质三维结构方面取得巨大飞跃,准确性可与冷冻电子显微术(又称冷冻电镜)(Cryo-EM)和X-射线晶体学等实验技术相媲美。
第一代AlphaFold依托蛋白质数据库PDB作为训练数据集,构建神经网络,采用深度学习预测氨基酸残基间的方向和距离,混合传统算法Rosetta对蛋白质结构进行同源建模、结构优化;与此不同的是,第二代AlphaFold则将折叠蛋白质视为“空间图”,基于神经网络系统进行“端到端”的训练,使用了进化相关的氨基酸序列,多序列比对以及对氨基酸对的评估来优化结构预测。研究人员使用蛋白质数据库中接近17万个不同的蛋白质结构,通过不断地迭代,AlphaFold系统学习到了基于氨基酸序列精确预测蛋白结构的能力。这一基于原子坐标近乎“暴力”的算法是全新的途径,是全新算法与强大算力的强强联合。
正如马里兰大学帕克分校计算生物学家,CASP共同创始人John Moult所言,从某种程度上而言,结构预测问题得到了解决。根据氨基酸序列准确预测蛋白质结构的能力将对生命科学和医学带来巨大的好处。这将极大地加快对细胞组成模块的理解,对于更快更先进的药物发现显然有很大帮助。Nature使用“它将会改变一切”来报道这一关键成果,研究人员之间也衍生出了“结构生物学家要失业了”的调侃。
然而,事实上,结构生物学研究或许反而会更上一层楼。AlphaFold部分结构预测的精度确实可与实验测定方法相媲美,这其中也不乏复杂超长的结构。但生物体环境是复杂的,体内蛋白质折叠结构受到更加复杂因素的影响,其使用的数据集也多来自于过去的静态结构测定方法;并且AlphaFold目前预测复杂蛋白复合物结构的能力还十分有限,在体内,最低能量状态则并非唯一的决定因素;而实验测定方法的确会受到蛋白质生物物理特性的局限导致无法获得蛋白复合物的完整结构,比如难以结晶的膜蛋白,但“眼见为实”为功能机制理解、药物开发、蛋白设计提供了扎实的实验证据,是不可缺失的一环。因此,AlphaFold的结构预测手段和实验测定手段可以相辅相成,成为实验科学家更有力的手段,共赢大于竞争。正如《返朴》总编、结构生物学家颜宁及其他大咖对此的深刻评议与发言:结构生物学的主语是生物学,是理解生命、是为了回答问题,做出生物学发现。
AlphaFold的表现证明了AI在辅助基础科学发现方面惊人的潜力。但“改变一切”过于乐观,也言之尚早,AlphaFold目前还不能预测复杂的分子机器,蛋白-蛋白相互作用非常复杂,存在极多的可能性。实验手段所揭示出来的蛋白-蛋白相互作用方式也还只是冰山一角,更何况在不同生理条件和过程中的结构变化。AlphaFold确实可以加快可成药靶点配体的发现速度,但对于新药发现而言,基于结构寻找合适配体只是第一步,先导化合物的选择性、过膜性、稳定性等成药性质的优化仍然需要大量的工作。
正如DeepMind团队在公司发布的博文中表示,他们相信,AI将成为人类拓展科学知识前沿最有力的工具之一!我们也相信,并且十分期待AlphaFold更加完美的表现!
?
参考链接:
1. 网页链接
2.网页链接
3.网页链接
4.药明康德《解决生物学50年来的重大挑战!生物界“AlphaGo”精准预测蛋白质结构》
5.返朴《颜宁等点评:AI精准预测蛋白质结构,结构生物学何去何从?》




 添加微信免费咨询
添加微信免费咨询
